CRISPR-CAS9 como ferramenta para edição do gene IT-15 na Doença de Huntington
Barra lateral de artigos
Conteúdo do artigo principal
Resumo
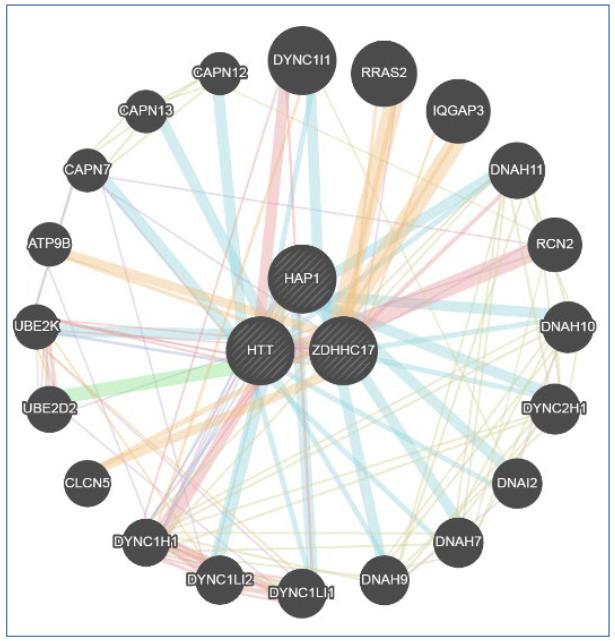
A Doença de Huntington (DH) é uma doença neurodegenerativa, autossômica dominante e hereditária que ocorre devido a uma mutação genética que gera uma sequência repetitiva de trinucleotídeos CAG, presentes no gene IT-15, gene da huntingtina, localizado no cromossomo 4. O objetivo foi revisar a neuropatologia da doença de Huntington (DH) e a utilização do método CRISPR-Cas9 para silenciar o gene IT-15 e verificar assim, a consequência nos genes HIP14 e HAP1, que possuem interação com a Huntigtina mutada e o resultado desta no organismo do paciente. Foram pesquisados artigos em bases indexadas (Scielo, PubMed e LILACs) com os seguintes descritores: ((Huntington) OR (Proteína Huntingtina)) AND (edição gênica). Também foi utilizada a ferramenta on line GeneMania, acesso livre, para análise de probabilidades e interações gênicas. O silenciamento do gene IT-15 acarreta alterações nas proteínas que interagem com a Huntingtina mutada, levando a perturbações em diversos processos.
Detalhes do artigo
Os autores mantêm os direitos autorais e concedem ao HSJ o direito de primeira publicação. A partir de 2024, as publicações serão licenciadas sob a Attribution 4.0 International 
 , permitindo seu compartilhamento, reconhecendo a autoria e publicação inicial nesta revista.
, permitindo seu compartilhamento, reconhecendo a autoria e publicação inicial nesta revista.
Os autores estão autorizados a assumir contratos adicionais separadamente para distribuição não exclusiva da versão do trabalho publicada nesta revista (por exemplo, publicação em repositório institucional ou como capítulo de livro), com reconhecimento de autoria e publicação inicial nesta revista.
Os autores são incentivados a publicar e distribuir seu trabalho on-line (por exemplo, em repositórios institucionais ou em sua página pessoal) a qualquer momento após o processo editorial.
Além disso, o AUTOR fica informado e consente que o HSJ possa incorporar seu artigo em bases de dados e indexadores científicos existentes ou futuros, nas condições definidas por estes a cada momento, o que envolverá, pelo menos, a possibilidade de que os titulares de esses bancos de dados podem executar as seguintes ações no artigo.
Referências
van der Burg JM, Björkqvist M, Brundin P. Beyond the brain: widespread pathology in Huntington's disease. Lancet Neurol. 2009;8(8):765-74. https://doi.org/10.1016/S1474-4422(09)70178-4
Martelli A. Aspectos clínicos e fisiopatológicos da doença de Huntington. Arch Health Invest [Internet]. 2014 [cited 2020 22 Sep];3(4):32-39. Avaiable from: http://www.archhealthinvestigation.com.br/ArcHI/article/view/687
Gil-Mohapel JM, Rego AC. Doença de Huntington: uma revisão dos aspectos fisiopatológicos. Rev Neurocienc 2011;19(4):724-34. https://doi.org/10.34024/rnc.2011.v19.8332
Shannon KM. Recent Advances in the Treatment of Huntington's Disease: Targeting DNA and RNA. CNS Drugs. 2020;34(3):219-228. https://doi.org/10.1007/s40263-019-00695-3 PMid:31933283
Intrieri ACU, Filho HB, Sabino MRLS, Ismail M, Furtado CC. Huntington: distúrbio no cromossomo 4. Rev UNILUS Ens Pesq [Internet]. 2015 [cited 2020 Sep 22];12(29):22-34. Avaiable from: http://revista.lusiada.br/index.php/ruep/article/view/687
Kaltenbach LS, Romero E, Becklin RR, et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007;3(5):e82. https://doi.org/10.1371/journal.pgen.0030082 PMid:17500595 PMCid:PMC1866352
Kolli N, Lu M, Maiti P, Rossignol J, Dunbar GL. CRISPR-Cas9 Mediated Gene-Silencing of the Mutant Huntingtin Gene in an In Vitro Model of Huntington's Disease. Int J Mol Sci. 2017;18(4):754. https://doi.org/10.3390/ijms18040754 PMid:28368337 PMCid:PMC5412339
Gonçalves GAR, Paiva RMA. Terapia gênica: avanços, desafios e perspectivas. Einstein (São Paulo) [Internet]. 2017;15(3):369-75. https://doi.org/10.1590/s1679-45082017rb4024 PMid:29091160 PMCid:PMC5823056
Warde-Farley D, Donaldson SL, Comes O, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010; 38(Suppl 2):W214-20. https://doi.org/10.1093/nar/gkq537 PMid:20576703 PMCid:PMC2896186
Saudou F, Humbert S. The biology of Huntingtin. Neuron. 2016;89(5):910-26. https://doi.org/10.1016/j.neuron.2016.02.003 PMid:26938440
ABH: Associação Brasil Huntington [Internet site]. Contexto genético da DH [access 2020 Nov 12]. Avaiable from: http://abh.org.br/o-que-e-doenca-de-huntington/contexto-genetico-da-dh/
A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72(6):971-83. https://doi.org/10.1016/0092-8674(93)90585-E
Gusella JA, MacDonald ME. Huntington's Disease: seeing the pathogenic process through a genetic lens. Trends Biochem Sci. 2006;31(9):533-40. https://doi.org/10.1186/gm80 PMid:19725930 PMCid:PMC2768966
Landles C, Bates GP. Huntingtin and the molecular pathogenesis of Huntington's Disease. EMBO Rep. 2004;5:958-63. https://doi.org/10.1038/sj.embor.7400250 PMid:15459747 PMCid:PMC1299150
Spitz M. Doença de Huntington e outras coréias. Rev Hosp Universitário Pedro Ernesto. 2010;9(1):29-38.
Frank S. Tetrabenazine: the first approved drug for the treatment of chorea in US patients with Huntington disease. Neuropsychiatric disease and treatment. 2010;6(1):657-65. https://doi.org/10.2147/NDT.S6430 PMid:20957126 PMCid:PMC2951749
Nance MA. Therapy in Huntington's Disease: where are we? Curr Neurol Neurosci Rep. 2012;12(4):359-66. https://doi.org/10.1007/s11910-012-0277-4 PMid:22544535
Shin JW, Kim KH, Chao MJ, et al. Permanent inactivation of Huntington's disease mutation by personalized allele-specific CRISPR/Cas9. Human Molecular Genetics. 2016;25(20):4566-76. https://doi.org/10.1093/hmg/ddw286 PMid:28172889 PMCid:PMC6078600
Ravache M, Abou-Sleymane G, Trottier Y. Neurodegenerative polyglutamine expansion diseases: physiopathology and therapeutic strategies. Pathol Biol (Paris). 2010;58(5):357-66. https://doi.org/10.1016/j.patbio.2009.12.004 PMid:20299163
Sun Y, Savanenin A, Reddy PH, Liu YF. Polyglutamine-expanded huntingtin promotes sensitization of n-methyl-d-aspartate receptors via post-synaptic density 95. J Biol Chem. 2001;276(27):24713-8. https://doi.org/10.1074/jbc.M103501200 PMid:11319238
Li XJ, Li SH, Sharp AH, et al. Huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995;378(6555):398-402. https://doi.org/10.1038/378398a0 PMid:7477378
Arend MC, Pereira JO, Markorski MM. O Sistema CRISPR/Cas9 e a possibilidade de edição genômica para a cardiologia. Arq Bras Cardiol. 2017;108(1):81-3. https://dx.doi.org/10.5935/abc.20160200
Yang W, Tu Z, Sun Q, Li XJ. CRISPR/Cas9: Implications for modeling and therapy of neurodegenerative diseases. Front Mol Neurosci. 2016;9(30):1-4. https://doi.org/10.3389/fnmol.2016.00030
Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. https://doi.org/10.7554/eLife.00471 PMid:23386978 PMCid:PMC3557905
Savic N, Schwank G. Advances in therapeutic CRISPR/Cas9 genome editing. Transl Res. 2016;168:15-21. https://doi.org/10.1016/j.trsl.2015.09.008 PMid:26470680
Singaraja RR, Huang K, Sanders SS, et al. Altered palmitoylation and neuropathological deficits in mice lacking HIP14. Hum Mol Genet. 2011;20(20):3899-909. https://doi.org/10.1093/hmg/ddr308 PMid:21775500 PMCid:PMC3177655